Со времени исследований О. Минковского (O. Minkowski) и Дж. Меринга (J. Von Mering) (1889-1892), которые вызывали диабет у собак путем удаления у них поджелудочной железы, установлено что недостаточность секреции инсулина является одним из факторов, необходимых для развития диабета. Однако механизм инсулиновой недостаточности при ИЗД и ИНЗД различен. При ИЗД масса и размеры поджелудочной железы меньше, чем у лиц, не больных диабетом или страдающих ИНЗД. Количество и объем островков у больных диабетом I типа, а также количество инсулина, экстрагируемого из поджелудочной железы, у этих больных снижено по сравнению с нормой. Некоторые авторы называют такие островки “атрофическими”, так как в них количество b-клеток резко уменьшено. С помощью иммуноцитохимических методов показано, что такие островки состоят из a-клеток (70%) и d-клеток (около 30%), причем a- и d-клетки обнаруживаются и в ацинозной ткани железы. Наряду с такими островками в поджелудочной железе больных ИЗД встречаются отдельные островки, в которых выявляются признаки гиперплазии b-клеток и признаки, указывающие на повышение активности оставшихся b-клеток – дегрануляция и увеличение ядра клеток. Однако количество b-клеток составляет менее 10% от общего их количества, выявляемого в поджелудочной железе практически здоровых лиц.
При ИНЗД поджелудочная железа и ее островковый аппарат практически не отличаются от таковых у лиц соответствующего возраста без нарушения углеводного обмена. Островки менее компактны, выявляется дольчатость железы вследствие развития фиброзной ткани, может быть незначительное снижение числа b-клеток, однако соотношение a-, b-, d- и РР-клеток не изменено.
Почти во всех случаях в поджелудочной железе обнаруживаются явления гиалиноза, что крайне редко встречается при ИЗД. Признаком функциональной недостаточности b-клеток при ИНЗД является уменьшение размера ядер клеток, комплекса Гольджи и эндоплазматической сети, что, вероятнее всего, связано с развитием склероза сосудов, фиброза и гиалиноза островков. Эти изменения выявляются в большей или меньшей степени в поджелудочной железе лиц пожилого возраста при отсутствии нарушения углеводного обмена. Недостаточность b-клеток при ИНЗД может быть следствием снижения чувствительности этих клеток к глюкозе (недостаток глюкорецептора), нарушения процессов вхождения кальция в В-клетку, его связывания с кальмодулином, изменения микротубулярно-микроворсинчатой системы, дефекта фибриллярных белков цитоплазмы.
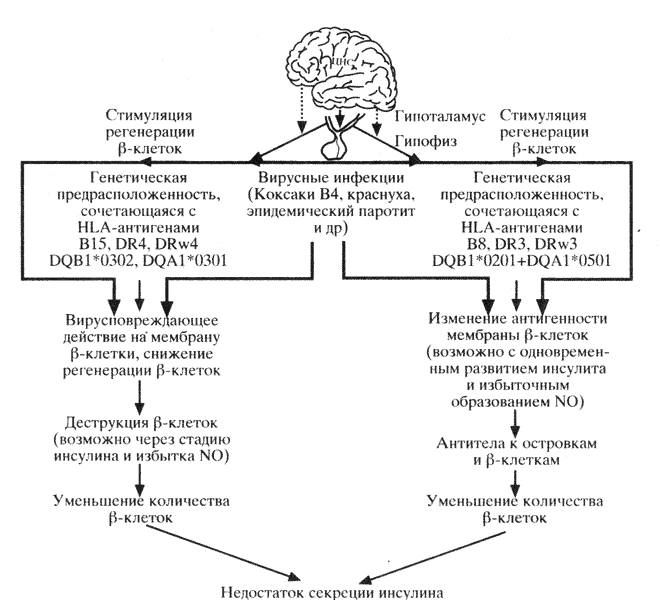
Диабет I типа в зависимости от механизма развития можно подразделить на два подтипа: аутоиммунный и вирусиндуцируемый. Механизмы патогенеза ИЗД представлены на схеме 31.
Аутоиммунный диабет характеризуется наличием признаков клеточно-опосредованного нарушения поджелудочной железы. Антитела определяются еще до развития клинических признаков диабета и поддерживаются на высоком уровне в течение нескольких лет после начала диабета, также частым сочетанием с прочими аутоиммунными эндокринопатиями и наличием антител к другим эндокринным железам (щитовидная железа, надпочечники и др.). Аутоиммунный диабет сочетается с антигенами системы HLA: B8, DR, DRw3, Dw3. При ИЗД четко выявляется ассоциация DR3 и генов локуса DQ (DQB1*0201+DQA1*0501). Причем указанные ассоциации генов локуса DR и DQ выявляются у лиц многих рас, за исключением японцев, где подобные ассоциации аллелей встречаются исключительно редко. Развивается в любом возрасте и чаще у лиц женского пола.
Для вирусиндуцированного диабета характерно лишь кратковременное образование антител к антигенам островков поджелудочной железы, которые, как правило, исчезают в течение года. Этот тип диабета не сочетается с аутоиммунными поражениями других эндокринных органов, развивается в более молодом возрасте (не старше 30 лет), чем аутоиммунный тип, и одинаково часто встречается как у мужчин, так и у женщин.
У больных, страдающих вирусиндуцированным типом диабета, выявляются антигены системы HLA: B15, DRw4, Dw4, DR4 и дополнительные аллели Cw3. Гены локуса DR4 ассоциируются с генами локуса DQ (DQB1*0302+DQA1*0301). Выше указано, что у больных этой группы антитела к антигенам островков поджелудочной железы выявляются значительно реже и время их циркуляции более ограничено. Однако характерной особенностью этих больных является повышенная наклонность к образованию антител к экзогенному инсулину.
Существует и третья форма ИЗД, которая сочетается с антигенами системы HLA: B8, DR3/B15-DR4 и соответствующих генов локуса DQ (DQA1*0501, *0301+DQB1*0201, 0302). В его генезе играют роль как аутоимммунные нарушения, так и вирусная инфекция. Заболевание развивается в детском возрасте, характеризуется почти тотальным поражением b-клеток, что сопровождается низким содержанием С-пептида в сыворотке крови.
В последние годы показано наличие еще одной – медленно прогрессирующей – формы сахарного диабета I типа, которая характеризуется медленным развитием инсулиновой недостаточности. Больные, страдающие этой формой, в течение 1-3 лет могут соблюдать диету и применять пероральные сахароснижающие препараты, что позволяет компенсировать нарушение углеводного обмена. Однако в последующем возникает резистентность к этим препаратам и для компенсации нарушения углеводного обмена больных переводят на инсулинотерапию. У таких больных в течение всего заболевания в сыворотке крови выявляются антитела к антигенам островков поджелудочной железы и отмечается прогрессивное снижение остаточной секреции.
Таким образом, независимо от инициирующих факторов и начальных механизмов диабета (вирусиндуцированный, аутоиммунный или медленно прогрессирующий) на последующих этапах в островках поджелудочной железы наблюдается деструкция и прогрессирующее уменьшение количества b-клеток вплоть до полного их исчезновения и развития абсолютной инсулиновой недостаточности.
Механизм деструкции b-клеток поджелудочной железы также сложен и длительное время активно обсуждались три модели патогенеза ИЗД: Копенгагенская модель (J. Nerup и соавт., 1989), Лондонская модель (G. Bottazzo и соавт., 1986), Стенфордская модель (H. McDevitt и соавт., 1987).
Последовательность патогенетических процессов в Копенгагенской модели была представлена следующим образом: а) инициация и деструкция b-клеток зависит от высвобождения под влиянием различных веществ (вирусы, химические вещества, избыток интерлейкина-1) антигена b-клеток; б) триггирование синтеза и высвобождения из макрофагов цитокина интерлейкина -1 (Ил-1); в)стимуляция секреции лимфокинов (g-интерферон и фактор некроза опухолей) Т-хельперами под влиянием Ил-1; г) g-интерферон и фактор некроза опухолей непосредственно принимают участие в разрушении b-клеток; причем g-интерферон индуцирует экспрессию генов HLA-II класса на клетках эндотелия капилляров, а Ил-1 увеличивает проницаемость капилляров и индуцирует экспрессию генов HLA-I и II класса в островке поджелудочной железы, что и замыкает “порочный круг“ деструкции новых b-клеток.
В Лондонской модели акцент отводился индукции генов HLA-II класса на эндокринных клетках островка поджелудочной железы под влиянием фактора некроза опухолей и g-интерферона при высокой концентрации Ил-1. Далее следует аберрантная экспрессия генов DR3 и DR4. При этих условиях Т-цитотоксические лимфоциты способствуют вместе с генами HLA-I класса усилению b-клеточного киллерового механизма (макрофаг/естественный киллер).
McDevitt с сотр.(1986) первыми секвестрировали локус DQ и показали, что в b-цепи этого локуса у здоровых лиц в 57-м положении находится аспарагиновая кислота, тогда как у больных диабетом она замещена на другие аминокислоты – валин, серин или аланин.
Значительным прорывом в понимании механизмов деструкции b-клеток поджелудочной железы явились исследования биологической значимости оксида азота (NO). Оксид азота является относительно стабильным свободным радикалом, период полужизни которого составляет несколько секунд. Продуктами окисления оксида азота являются нитраты и нитриты. Длительное время о месте образования и биологической функции оксида азота судили косвенно по экспрессии NO-синтазы. Оказалось, что в организме имеется несколько изоформ NO-синтазы, две из которых называются основными (эндотелиальная и нейрональная) и третья изоформа – индуцированная (иNO-синтаза).
Исходным продуктом для образования оксида азота является L-аргинин. Последний под воздействием NO-синтазы превращается в L-цитрулин и оксид азота. В случае взаимодействия L-аргинина с эндотелиальной или нейрональной NO-синтазой образующийся оксид азота участвует в процессах передачи различных сигналов (в нервной и других системах) или оказывает прямое влияние на тонус периферических сосудов. Уникальной функцией (цитотоксическая и цитостатическая) обладает оксид азота, образующийся под воздействием индуцированной NO-синтазы. Имеется несколько селективных ингибиторов NO-синтаз. Один из них – аминогуанидин практически селективно угнетает иNO-синтазу (в 40 раз сильнее ингибирует иNO-синтазу по сравнению с основными NO-синтазами).
Исследованиями последних лет показано, что именно оксид азоту, который образуется в островках и b-клетках поджелудочной железы, принадлежит основная роль в механизмах разрушения и гибели b-клеток, что и приводит к резкому уменьшению их количества и развитию клинического ИЗД.
Считаем необходимым несколько подробнее остановиться на этапах механизма, оперирующего в b-клетках и приводящих к аутоиммунному диабету.
Островок поджелудочной железы, как известно, содержит около 2000 эндокринных клеток, из которых 65% приходится на b-клетки и 30% – a-клетки, и неэндокринных клеток (около 10 макрофагов на островок, фибробласты, эндотелиальные и дендрические клетки). Неэндокринные клетки островка опосредуют лизис островковых клеток, наблюдаемый при обработке интактных островков Ил-1 (L. Bergmann и соавт., 1992). Макрофагам принадлежит центральная роль в инициации каскада иммунных реакций, приводящих в конечном итоге к аутоиммунной деструкции b-клеток. Активирование макрофагов осуществляется несколькими путями: взаимодействием с липополисахаридами, g-интерфероном или фактором некроза опухолей, что сопровождается высвобождением большого количества оксида азота и цитокинов (Ил-1),которые индуцируют экспресиию иNO-синтазы, а последняя в свою очередь оперирует уже непосредственно в b-клетках, способствуя образованию оксида азота из L-аргинина. Образование оксида азота сопровождается значительным повышением нитритов, которые по мнению многих исследователей также участвуют в механизмах повреждения b-клеток.
Оксид азота, образуемый макрофагами, функционирует как специфическая эффекторная молекула, но может также участвовать в деструкции b-клеток. Однако основное повреждающее действие оказывает оксид азота, который, как указано выше, образуется непосредственно в b-клетке. Так, J. Corbett и соавт. (1995) показали, что экспрессия иNO-синтазы в макрофагах и образование ими оксида азота практически не влияет на функцию островка, тогда как экспрессия иNO-синтазы в b-клетках и образование здесь оксида азота полностью угнетает секрецию инсулина.
Экспрессия иNO-синтазы в b-клетках индуцируется Ил-1, который связывается с соответствующим рецептором, локализованным на мембране b-клетки. Комплексирование Ил-1 со своим рецептором на b-клетке происходит при участии специфического белка-”белка антагониста рецептора Ил-1”. В регуляцию экспрессии мРНК иNO-синтазы вовлечены транскрипционные регуляторы c-fos, c-jun и ядерный фактор- NF-kb. Установлено также, что ген иNO-синтазы локализуется на 11-й хромосоме в непосредственной близости от “полиморфной диабетической области” (область гена, кодирующего синтез инсулина), что позволяет предполагать вовлечение этих двух областей 11-й хромосомы в патогенез ИЗД.
Помимо оксида азота, значительное место в механизмах деструкции b-клеток отводится простагландинам островка поджелудочной железы. Инфильтрация островка лимфоцитами и макрофагами (инсулит) постоянно встречается на самых ранних этапах развития диабета. Клеточные элементы, участвующие в “инсулите”, индуцируют экспрессию цитокинов (Ил-1 и др.), которые в свою очередь способствуют как экспрессии иNO-cинтазы и последующему образованию свободно-радикального соединения, которым является оксид азота, так и циклооксигеназы, ответственной за биосинтез из арахидоновой кислоты провоспалительных простагландинов, усиливающих инсулит и приводящих вместе с оксидом азота к аутоиммунной деструкции b-клеток.
Следует отметить, что при исследованиях на NOD мышах (экспериментальная модель аутоиммунного диабета) установлено, что в период развертывания аутоиммунного диабета четко прослеживается две фазы процесса: NO-зависимая и независимая. Оксид азота участвует в механизмах развития диабета на ранней стадии лимфатической инфильтрации островка поджелудочной железы. Накопление большого количества лимфоцитов повышает Т-лимфоцитопосредованное повреждение b-клеток независимо от образования NO.
Помимо аминогуанидина, способностью к ингибированию экспрессии NO-синтазы, а следовательно и образованию оксида азота, обладает никотинамид, который применяется в клинике для лечения манифестных форм ИЗД. С 1994 г. проводится многоцентровое европейское исследование по изучению влияния никотинамида на профилактику и лечение ИЗД, результаты которого будут представлены не ранее 1998 г. Перспективными для применения с целью профилактики диабета является упомянутый аминогуанидин.
ИЗД представляет собой гетерогенную группу. Выделяют по крайней мере 3 подгруппы сахарного диабета I типа: аутоиммунный, вирусиндуцируемый и медленнопрогрессирующий. Для аутоиммунного подтипа характерно наличие антител к островкам поджеледочной железы, которые, как правило, выявляются за несколько месяцев и даже лет до манифестации ИЗД. Сахарный диабет у таких больных возникает в любом возрасте (чаще у лиц женского пола) и сочетается с другими эндокринными аутоиммунными заболеваниями. У этих больных одновременно выявляются антитела к антигенам клеток других эндокринных органов и тканей.
При вирусиндуцированном типе сахарного диабета антитела к островкам поджелудочной железы непостоянны и исчезают уже через 1 год от начала заболевания. Заболевание, как правило, начинается остро, нередко с кетоацидоза или комы, встречается в молодом возрасте (до 30 лет) одинаково часто у мужчин и женщин в отсутствие других аутоиммунных заболеваний.
Медленно прогрессирующий тип ИЗД характеризуется медленным началом, в более зрелом возрасте, в течение нескольких лет имеет благоприятное течение, т.е. сначала состояние углеводного обмена компенсируется диетой, затем – пероральными сахароснижающими препаратами и, наконец, выявляется инсулинозависимость. При определении аутоантител к генам островка поджелудочной железы уже изначально определяются один или несколько типов таких антител в сыворотке крови больных. Кроме того, параллельно прогрессивному снижению эффективности действия пероральных сахароснижающих препаратов при обследовании у больных выявляют низкое содержание С-пептида в сыворотке крови, и компенсация углеводного обмена достигается только инсулинотерапией.
Таким образом, независимо от начальных механизмов (вирусиндуцируемый, аутоиммунный или медленно прогрессирующий) деструкции b-клеток на последующих стадиях процесса происходит уменьшение их количества вплоть почти до полного исчезновения b-клеток с развитием абсолютной инсулиновой недостаточности.
Согласно G. Eisenbarth (1986), патогенез ИЗД можно разделить на 6 стадий, медленно прогрессирующих и переходящих одна в другую: 1) генетическая предрасположенность (обусловленная наличием определенных гаплотипов генов HLA-системы I,II и III класса); 2) триггирование или инициация иммунных процессов; 3) стадия активных иммунологических процессов; 4) прогрессивное снижение первой фазы секреции инсулина, стимулированной внутривенным введением глюкозы; 5) клинически явный или манифестный диабет; 6) полная деструкция b-клеток.
Патогенез ИНЗД отличается от описанного выше (схема 32).
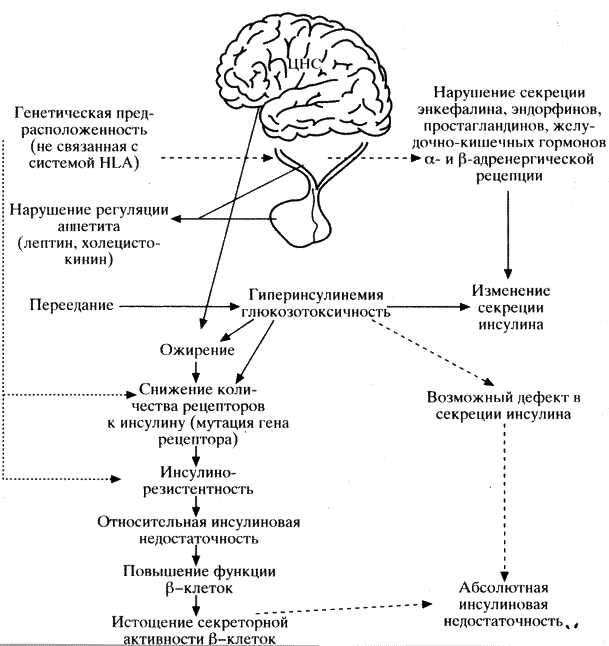
Генетическая предрасположенность при диабете этого типа играет более значительную роль, чем при ИЗД. Разрешающими факторами могут служить ожирение и беременность. Показано, что у больных, страдающих ожирением, похудание приводит к снижению исходной концентрации глюкозы и инсулина в ответ на прием пищи. Возврат больных к избыточному питанию вновь сопровождается гипергликемией и гиперинсулинемией натощак, а также ухудшением секреции инсулина в ответ на прием пищи.
Новые данные получены и по патогенезу ИНЗД. Так, многочисленными исследованиями установлено, что развитие диабета II типа обусловлено инсулинорезистентностью и нарушением функции b-клеток. Соотношение этих двух компонентов патогенеза ИНЗД различно как в отдельных популяциях, так и у конкретных больных одной популяции. Неясно также, какой из двух перечисленных дефектов является первичным при ИНЗД. Так, у индейцев племени пима инсулинорезистентность предшествует ИНЗД. У родственников первой степени родства больных диабетом II типа в период, когда имеется ещё нормальная толерантность к глюкозе, при обследовании уже выявляется снижение чувствительности к инсулину в мышцах при наличии гиперинсулинемии. В то же время у больных ИНЗД, имеющих нормальную или слегка сниженную массу тела, на ранних стадиях заболевания имеет место инсулинопения.
Причины инсулинорезистентности при ИНЗД гетерогенны. Несмотря на то, что полностью её механизмы не раскрыты, тем не менее за последние годы мы намного продвинулись вперед в понимании этиологических и других причин, приводящих к инсулинорезистентности при диабете II типа. В развитии инсулинорезистентности чётко прослеживается два компонента: генетический, или наследственный, и приобретенный. Родственники первой степени родства с нарушенной и даже с нормальной толерантностью к глюкозе имеют выраженную инсулинорезистентность по сравнению с лицами контрольной группы. У монозиготных близнецов с ИНЗД инсулиновая резистентность также более выражена по сравнению с близнецами без диабета. Приобретенный компонент инсулинорезистентности проявляется уже в период манифестации диабета. В ряде исследований показано, что имеющаяся умеренная инсулинорезистентность у родственников первой степени родства при сохранении нормальной толерантности к глюкозе значительно усугубляется при нарушении у них углеводного обмена. Аналогичные данные получены при проведении исследований у монозиготных близнецов.
В настоящее время клиническими и экспериментальными исследованиями показано, что одной из причин проявления инсулинорезистентности в более выраженной степени является глюкозотоксичность, т.е. состояние длительной гипергликемии. Помимо этого, глюкозотоксичность способствует десенситизации b-клеток, что проявляется ухудшением их секреторной активности. Установлено также, что некоторые аминокислоты и, в частности, глютамин значительно влияют на действие инсулина, модулируя поглощение глюкозы. Наблюдаемая в этих случаях десенситизация является следствием образования продуктов обмена гексозаминов (гексозаминовый шунт). Глютамин и фермент фруктозо-6-фосфат аминотрансфераза необходимы для конверсии фруктозо-6-фосфата в глюкозамин-6-фосфат и для нормального функционирования этого шунта.
Свободные жирные кислоты оказывают ингибирующее влияние на окисление глюкозы (цикл Рэндла) и участвуют в поддержании и усилении состояния инсулинорезистентности. Нарушение пульсирующей секреции инсулина и его влияния на распределение глюкозы в организме также является фактором, способствующим инсулинорезистентности. При ИНЗД также имеет место нарушение инсулинорецепторного взаимодействия (уменьшение количества рецепторов к инсулину, снижение их аффинности), что сопровождается усилением клинических проявлений инсулинорезистентности и восстанавливается почти до нормы при снижении массы тела.
Помимо рецепторных, имеется значительное число пострецепторных механизмов, участвующих как в механизмах инсулинорезистенности, так и в механизмах развития диабета. Инициация передачи гормонального сигнала начинается с фосфорилирования b-субъединицы инсулинового рецептора, которое осуществляется тирозинкиназой. Это фосфорилирование, а затем поддерживающееся аутофосфорилирование рецептора инсулина необходимо для последующих этапов пострецепторного действия инсулина и, в частности, для активирования и транслокации глюкозных транспортеров (ГЛЮТ), наиболее важным из которых является ГЛЮТ-4. Экспрессия этого транспортера имеет место только в скелетных мышцах, мыщцах сердца и жировой ткани. Гликозилирование или уменьшение транслокации ГЛЮТ-4 сопровождается инсулиновой резистентностью.
Причиной инсулинорезистентности может быть мутация гена инсулинового рецептора. По мнению S. Taylor и D. Moller (1993), мутации инсулинового рецептора следует подразделять на V классов:1) мутации, приводящие к снижению скорости биосинтеза рецептора; 2)мутации, ухудшающие внутриклеточный транспорт и посттрансляционный процессинг ; 3) мутации, приводящие к дефектам связывания инсулина; 4) мутации, сопровождающиеся снижением рецепторой активности тирозинкиназы и 5) мутации, ускоряющие деградацию инсулинового рецептора. К I классу мутаций относятся бессмысленные мутации кодона 897, кодона 672 гена рецептора инсулина, сопровождающиеся значительным снижением уровня мРНК гена инсулинового рецептора. Выявлено более 30 точечных мутаций гена инсулинового рецептора, в том числе относящихся ко II классу и идентифицированных при различных формах диабета, включая ИНЗД, сопровождающихся инсулиновой резистентностью. Несколько мутантных рецепторов характеризуются дефектами посттрансляционной модификации. При этом такая мутация может сопровождаться дефектом транспорта рецептора к клеточной поверхности, снижением аффинности рецептора или никак не отражаться на функциональной активности рецептора. Среди описанных мутаций III класса следует отметить две мутации инсулинового рецептора, сопровождающиеся снижением способности связывания рецептора с инсулином (снижение аффинности) и мутацию, приводящую к повышению аффинности инсулинового рецептора. К IV классу мутаций относятся: а) мутации b-субъедиицы рецептора, приводящие к снижению инсулинстимулированной рецепторной тирозинкиназы (делеции экзона 17-22, мутации кодона 1109, мутации юкстамембранного домена, мутации, при которых резко снижается фосфорилирование IRS-1 или субстрата-1 инсулинрецепторной киназы и др.); б) мутации внеклеточного домена, также сопровождающиеся ингибированием тирозинкиназной активности; в) киназодефицитные мутации, сопровождающиеся снижением эндоцитоза инсулинрецепторного комплекса и нарушением обратной регуляции-”down – regulation”; г) киназодефицитные мутации, приводящие к инсулиновой резистентности. И, наконец, мутации глютамина460 (GLU460) относят к мутациям V класса, которые сопровождаются ускорением деградации инсулинового рецептора.
Жировая ткань является местом образования фактора, который ингибирует действие инсулина. S. Hotamisligil и B. Spiegelman (1994) установили, что таким веществом является a-фактор некроза опухолей. Инсулинорезистентность сопровождается повышением экспрессии в жировой ткани мРНК a-фактора некроза опухолей. Нейтрализация a-фактора некроза опухолей приводит к улучшению действия инсулина в скелетных мышцах и жировой ткани, тогда как в печени этого эффекта не наблюдается. B. A. Maddux и соавт. (1995) описали ещё один механизм инсулиновой резистентности при ИНЗД. Ими был идентифицирован белок, названный гликопротеин 1 плазматической мембраны (РС 1) с молекулярной массой 115-135 кДа, который снижает активность киназы рецептора к инсулину. Интересные данные, проливающие свет на механизмы наследования инсулиновой резистентности, получили C. Reynet и C. Kahn (1993). Они идентифицировали мРНК, которая избыточно экспрессируется в мышцах больных ИНЗД. Эта мРНК кодирует небольшой ГТФ-связывающий белок, названный ими rad (ras, сочетающийся с диабетом). Подобно ras белку rad белок связывает ГТФ (гуанидин трифосфат). Исследования в этой лаборатории (J. Zhu и соавт., 1995) показали, что rad является уникальным белком, внутриклеточный уровень которого повышается в мышцах больных диабетом, каким то образом участвуя в механизмах инсулинорезистентности. Следует отметить, что повышение rad белка выявляется лишь у части больных, страдающих сахарным диабетом II типа.
Как указывалось выше, обязательным компонентом патогенеза ИНЗД является нарушение функции b-клеток. Дисфункция b-клеток развивается как результат совместного воздействия нескольких факторов. Во-первых, это глюкозотоксичность, т.е. состояние длительной, хронической гипергликемии, которое приводит к снижению секреторного ответа b-клеток на стимуляцию повышенным уровнем глюкозы в крови. Это проявляется как снижением или полным отсутствием первой фазы секреции инсулина, так и нарушением пульсирующей секреции инсулина. Во-вторых, установлено, что при ИНЗД имеет место снижение массы b-клеток. Отмечается нарушение конверсии проинсулина в инсулин. У больных ИНЗД изменяется отношение проинсулина к интермедиатным формам инсулина (в сторону увеличения последних), которые, как и сам проинсулин, обладают лишь незначительной сахароснижающей активностью (приблизительно 5-10% по сравнению с инсулином). Показано также, что при диабете II типа снижается количество глюкозного транспортера II типа (ГЛЮТ-2), который является единственным транспортером глюкозы, оперирующим в b-клетках. Правда, у диабетических животных количество ГЛЮТ-2 уменьшалось лишь на 40-80%, что является достаточным для сохранения нормального поступления глюкозы в b-клетку. Однако, по данным A. Valera и соавт. (1994), трансгенные мыши с низким (до 80%) уровнем ГЛЮТ-2 становились диабетическими.
Одной из причин дисфункции b-клеток может быть нарушение глицерин-фосфатного шунта, который является важным сигнальным механизмом в глюкозостимулированной секреции инсулина. Снижение митохондриальной глицерин-фосфат дегидрогеназы выявлено как у экспериментальных диабетических животных, так и у больных ИНЗД.
Глюкокиназа является одним из важнейших ферментов, регулирующих метаболизм глюкозы в b-клетках. Мутации гена глюкокиназы выявляются почти у 50% больных, страдающих диабетом, так называемым MODY типом (диабет взрослого типа у молодежи). К настоящему времени описано более 20 различных точечных мутаций гена глюкокиназы.
Важным открытием последних лет явилась идентификация мутаций митохондриального гена как причина диабета. Митохондриальная ДНК, состоящая из 16569 пар оснований, кодирует 13 ферментов окислительного фосфорилирования. Её мутация обычно касается лейцина тРНК или так называемой мутации tRNALeu(UUR). Впервые такая точечная мутация митохондриальной ДНК была описана при MELAS cиндроме (митохондриальная миопатия, лактат ацидоз, энцефалопатия и инсультоподобные эпизоды), который часто встречается в Японии (A. Chomyn и соавт., 1992). Составляющей частью указанного синдрома является наличие ИЗД или ИНЗД с сенсорной потерей или без потери слуха (T. Kadowaki и соавт., 1994), которая, как правило, развивается после клинической манифестации диабета. Интересно, что ИЗД при этом синдроме (был проведен скрининг 55 лиц, имеющих явный ИЗД или указания в анамнезе о наличии его у других членов семьи) протекает по типу медленно прогрессирующего ИЗД, т.е. вначале диабет манифестируется в виде ИНЗД, а затем присоединяются симптомы инсулиновой недостаточности с наличием аутоантител к антигенам островка поджелудочной железы. Изучая 27 больных с таким медленно прогрессирующим типом ИЗД, Y. Oka и соавт. (1993) у 3 из них (11%) выявили митохондриальную точечную мутацию нуклеотида 3243 (tRNALeu(UUR)), тогда как у 50 здоровых лиц и 30 больных ИНЗД таковая мутация отсутствовала.
Среди других причин, влияющих на нарушение функции b-клеток при ИНЗД, следует отметить нарушение гена, кодирующего IRS-1 (субстрат-1 для инсулиновой рецепторной киназы); нарушение гена, локализованного на 4q хромосоме и кодирующего FABP-2 (2-й белок, связывающий жирные кислоты). Кроме того, имеются данные о том, что мутация гена гликогенсинтазы, точечные мутации гена b3-адренорецептора, точечные мутации 2-го экзона гена рецептора к глюкагону вовлечены в патогенез нарушения функции b-клеток и патогенез ИНЗД.
Хроническая гипергликемия снижает способность b-клеток отвечать секрецией инсулина на острую стимуляцию глюкозой. Такая развившаяся “слепота” b-клеток к глюкозе сопровождается нарушением первой и второй фазы секреции инсулина. Выявленные нарушения секреции инсулина при хронической гипергликемии полностью обратимы при нормализации углеводного обмена. Это явление, т.е. способность хронической гипергликемии нарушать секрецию инсулина, получило название токсичности глюкозы, или глюкозотоксичность.
Нарушение чувствительности b-клеток к глюкозе является следствием десенситизации, которая связана с нарушением активности фосфолипазы С, гидролиза мембранных фосфоинозитидов и уменьшением образования инозитолтрифосфата и диацилглицерина. Не исключается, что феномен десенситизации связан с повышенным образованием в островках поджелудочной железы простагландина Е2, ингибирующего секрецию инсулина.
Феномен глюкозотоксичности объясняет клинические наблюдения, показывающие, что нормализация уровня глюкозы у больных диабетом с помощью диеты, препаратов сульфонилмочевины или инсулина приводит к улучшению секреции инсулина, которая может поддерживаться в течение некоторого времени даже при перерыве в лечении.
Глюкозотоксичностью, видимо, объясняется феномен “медового периода” при ИЗД, когда у вновь выявленных больных сахарным диабетом I типа после начала инсулинотерапии и нормализации при этом углеводного обмена наблюдается улучшение течения диабета при снижении дозы или даже полной отмене инсулина. Теория глюкозотоксичности может объяснить в какой-то степени явление вторичной резистентности больных диабетом II типа к пероральной терапии препаратами сульфонилмочевины. Такая вторичная недостаточность может быть следствием десенситизации b-клеток к стимулирующему влиянию этих препаратов на секрецию инсулина. Нормализация углеводного обмена инсулинотерапией восстанавливает чувствительность b-клеток к глюкозе, и в дальнейшем такие больные могут быть переведены на лечение препаратами сульфонилмочевины.
Рядом исследований показано, что дериваты циклооксигеназы, главным образом простагландин Е2, угнетают ответ инсулина на стимуляцию глюкозой, тогда как производные липооксигеназы, главным образом 15-НРЕТЕ, стимулируют секрецию инсулина. Такие препараты, как колхицин или фуросемид, усиливающие синтез простагландинов, снижают секрецию инсулина, а нестероидные противовоспалительные препараты, главным образом салицилаты, угнетающие циклооксигеназу, повышают ответ инсулина на различные стимуляторы. Предполагают, что при ИНЗД повышается чувствительность к эндогенным простагландинам, что и является основной причиной нарушения секреции инсулина при этой патологии.
MODY-тип (сахарный диабет взрослого типа у молодежи, или “масонский тип” диабета) также является гетерогенным заболеванием. Молекулярно-генетические исследования показали наличие по крайней мере 3 синдромов: MODY-1 (сочетается с нарушениеми 20q хромосомы), MODY-2 (сочетается с различными мутациями гена глюкокиназы на 7р хромосоме) и MODY-3 (сочетается с нарушениями 12q хромосомы).
Выявлено несколько (около 10) семей в мире, у которых сахарный диабет II типа сочетался с различными мутациями гена инсулина, приведшими к различным замещениям аминокислот в молекуле инсулина (инсулин Чикаго- замещение в В25 фенилаланина на лейцин; инсулин Лос-Анджелес – замещение фенилаланина в В24 на серин; инсулин Вакаяма – замещение валина в А3 на лейцин) или нарушению структуры в молекуле проинсулина: в 2 случаях (проинсулин Токио и Бостон) аргинин в положении 65 был замещен на гистидин, что приводило к нарушению конверсии проинсулина в инсулин; и в одном случае (проинсулин Провиденс) выявлено замещение гистидина на аспарагиновую кислоту в положении В10, что также нарушает конверсию проинсулина в инсулин.
Представленные материалы еще раз подтверждают, что в патогенезе сахарного диабета любого типа участвуют одновременно несколько механизмов, степень выраженности которых различна при определенных типах указанной патологии.