Этиотропное и патогенетическое лечение, профилактика
Оглавление
ГЕНЕТИЧЕСКИЕ РАССТРОЙСТВА И УРОЛИТИАЗ -> Генетически детерминированные виды уролитиаза
Генетически детерминированные виды уролитиаза
Первичная гипероксалурия тип 1 (ПГ1) (МсК 259900) это аутосомно-рецессивное нарушение метаболизма глиоксалата вызванное недостаточностью в печени промежуточного метаболического фермента аланин-глиоксилат аминотрансферазы. Описана так же частая мутация, приводящая не к снижению активности, а к снижению компартментализации фермента (в митохондриях вместо пероксисом) G630A. Множество друтих специфичных мутаций было идентифицировано (Gly170Arg, Pro11Leu) и их широкое разнообразие изменило аспекты клинических подходов к ПГ1, особенно - к пренатальной диагностике. Клинически ПГ1 характеризуется повышением уровня экскреции оксалата и гликолата и хроническим накоплением кальция оксалата в мочевом тракте (уролитиаз и нефролитиаз) или почечной паренхиме (нефрокальциноз). ПГ1 очень гетерогенное состояние на клиническом и молекулярных уровнях. На клиническом уровне ПГ1 - есть заболевание почек, но на молекулярном уровне - это заболевание печени (Рис. 11.).
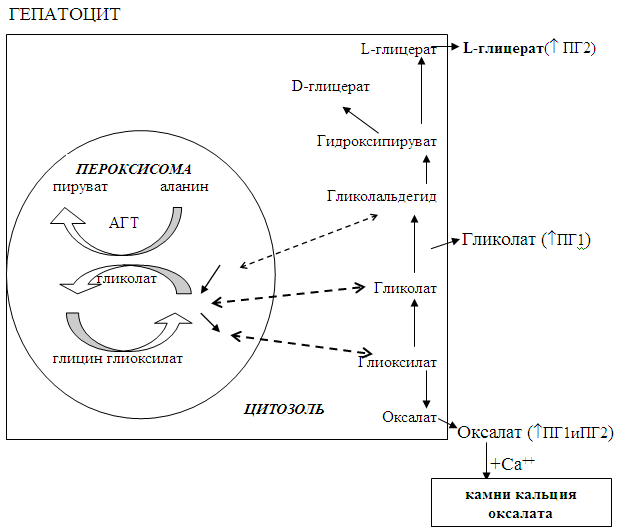
Рис. 11. Механизм патогенеза ПГ на уровне пероксисом гепатоцитов.
Клини фенотип болезни крайне гетерогенен. Некоторые авторы выделяли в данном заболевании неонатальные, детские и взрослые формы, представляющие на самом деле континуум различающихся по срокам и тяжести манифестации клинических фенотипов. Возраст начала болезни широко варьирует - от 1-го года жизни до 7-го десятилетия, в большинстве случаев - до 5 лет. У большинства пациентов заболевание манифестирует либо почечной коликой, либо асимптоматической выраженной гематурией, развивающейся вследствие оксалатного уролитиаза. Последний наряду с нефрокальцинозом неуклонно прогрессирует, приводя к развитию почечной недостаточности, которая переходит в уремию и ведет к летальному исходу в период до 20 лет у 80% больных. Вследствие почечной недостаточности кальций оксалат может накапливаться почти во всех тканях организма (системный оксалоз).
Наиболее эффективным методом каузальной терапии является пересадка печени, которую, как правило, сочетают с пересадкой почки, замещая биохимически и патофизиологически дефектные органы одновременно. В медицине накоплен опыт более 100 сочетанных трансплантаций печени и почек по поводу ПГ1. Частичная трансплантация печени клинически не эффективна. Изолированная трансплантация почек способствует улучшению на короткое время и при нормальных условиях не является методом лечения на длительный период, потому что не убирает причину заболевания. Коррекция метаболизма максимальна, если пересадка печени предпринята до формирования терминальной стадии почечной недостаточности, так что пересадки почки можно избежать. Диетарные ограничения и оксалата и кальция не эффективны. У некоторых больных назначение фармакологических доз пиридоксина, кофактора всех аминотрансфераз приводило к снижению экскреции оксалата. В литературе обсуждается возможность генотерапии данного заболевания.
Первичная гипероксалурия тип 2 (ПГ2) (МсК260000) ранее была в одной категории с ПГ1, потому что имеет много общих симптомов. ПГ2 имеет слабее выраженные симптомы, чем ПГ1, тем не менее некоторые пациенты достигают конечной стадии почечной недостаточности. Как и ПГ1 ПГ2 есть аутосомно-рецессивное заболевание, но только на биохимическом уровне.
ПГ2 вызывается недостатком промежуточного фермента глиоксилатредуктазы. У человека глиоксилатредуктаза способствует превращению глиоксилата в гликолат. При недостатке глиоксилатредуктазы больше глиоксилата может метаболизироваться в оксалат. Глиоксилат, будучи реактивной молекулой, токсичен для тканей, вызывая изменения ряда ферментных систем. Гликолат, напротив, выраженным токсическим действием не обладает.
Методы каузальной терапии включают гемодиализ и перитонеальный диализ (менее эффективно) [46].
Х-сцепленный рецессивный нефролитиаз (МсК 310468), Болезнь Дента (Dent's Disease) (МсК 300009), и возможно, Тип III гипофосфатемического рахита (МсК 307800) являются аллельными вариантами Х-сцепленного тубулярного почечного нарушения характеризующегося протеинурией (низкомолекулярный белок), гиперкальциурией, нефрокальцинозом, нефролитиазом и почечной недостаточностью. Почечные депозиты состоят из кальция фосфата и кальция оксалата. Накопленные данные показывают, что Х-сцепленный нефролитиаз, болезнь Дента и тип III гипофосфатемического рахита вызываются мутацией в CLCN5 гене Хр11.22 (МсК 300008).
Синдром Леша-Найхана (Lesch-Nyhan), (McK 308000) - это Х-сцепленное рецессивное заболевание, которое развивается в результате дефицита фермента гипоксантин-гуанин-фосфорибозилтрансферазы. Клинический фенотип гетерогенен. Различают три клинические формы: классическую или собственно болезнь Леша-Найхана, атипичную без поражения ЦНС, атипичную с поражением ЦНС. Помимо задержки умственного развития и неврологической симптоматики у этих пациентов часто развивается уролитиаз из-за повышения синтеза и экскреции мочевой кислоты. Аллопуринол, ингибитор ксантиноксидазы, снижает уровни сывороточной мочевой кислоты и предотвращает большинство симптомов, связанных с гиперурикемией, не оказывая влияния на неврологическую симптоматику. Наибольший терапевтический эффект связан с препаратами нейротрансмиттеров (L-5-гидрокситриптофан, КарбиДОПА, ЛеваДОПА, Тетрабеназин).
Повышение активности фосфорибозил-пирофосфат синтетазы - это Х-сцепленное рецессивное состояние, при котором повышается синтез пуринов, развивается подагра и мочекислый уролитиаз. Ген PRPS1, локализованный в Xq22-q24 (МсК 311850), и ген PRPS2 в Xp22.3-p22.2 (МсК 311860) ответственны за развитие данного состояния. Клинический фенотип гетерогенен: различают более тяжелую детскую и более легкую взрослую форму
Ксантинурия (МсК 278300) это аутосомно-рецессивное заболевание, при котором повышается мочевая экскреция ксантина и, в меньшей мере, гипоксантина. Эта болезнь чаще встречается у мужчин. Причиной болезни является дефицит фермента ксантин оксидазы (ксантин дегидрогеназы), который катализирует превращение гипоксантина в ксантин и далее в мочевую кислоту. Мутации в XDN гене, локализованном в хромосоме 2p22.3-p22.2 обеспечивают этот дефицит. Клинический фенотип гетерогенен. Различают классическую и кофактор-дефицитную формы. Классическая подразделяется на типы I и II, но только на биохимическом, а не на клиническом уровне.
Недостаточность аденин фосфорибозилтрансферазы (МсК 102600) является аутосомно-рецессивным заболеванием, проявляющимся повышением синтеза и мочевой экскреции 2,8-дигидроксиаденина, который является плохо растворимым соединением, легко кристаллизуется в моче и формирует камни. APRT ген, мутации которого обеспечивают данное состояние, локализован в хромосоме 16q22.2-23.2. В большинстве случаев заболевание ошибочно распознается как уратный литиаз на основании идентичной химической реактивности кристаллов мочевой кислоты и 2,8-дигидроксиаденина и рентгенпрозрачности обоих типов камней, что сопровождается назначением неадекватной терапии.
Цистинурия (МсК 220100) наследственное заболевание почек при котором повышается мочевая экскреция цистина, лизина, аргинина и орнитина. Цистин является плохо растворимым, и поэтому легко образуются мочевые конкременты. С генетической точки зрения цистинурия это гетерогенное заболевание, имеющее три варианта. Цистинурия тип 1 обусловлена мутацией в CSNU1 гене в 2q16.3 хромосоме, результатом которой является снижение резорбции цистина и других аминокислот. Цистинурия тип 2 и цистинурия тип 3 вызываются мутациями пока ещё не точно определённых генов, расположенных в хромосоме 19q13.1.
32-36
| Назад | Вперед |