Этиотропное и патогенетическое лечение, профилактика
Оглавление
КАЛЬЦИЕВЫЕ КАМНИ -> Почечный канальциевый ацидоз -> Проксимальный канальцевый ацидоз (тип II)
Проксимальный канальцевый ацидоз (тип II)
Проксимальный ПКА связан со снижением реабсорбции в проксимальных канальцах бикарбоната, что предположительно обусловлено недостаточной продукцией карбоангидразы. Вместо реабсорбции 85 % профильтрованного бикарбоната проксимальные канальцы могут реабсорбировать лишь 60 %, в результате чего в дистальные канальцы поступает 40 % профильтрованного объема вместо обычных 15 %. Поскольку в дистальных канальцах максимально может реабсорбироваться только 15 % профильтрованного объема бикарбоната, до 25 % его теряется с мочой. Проксимальный ПКА обычно протекает тяжелее дистального, т.к. неспособность к рабсорбции бикарбоната в дистальных канальцах (что бывает редко) приводит к потере лишь 15 % профильтрованного бикарбоната. При его выведении с мочой уровень его в сыворотке снижается до порогового, при котором прекращается его экскреция с мочой. При этом уровне (15 - 18 ммоль/л) количество профильтрованного бикарбоната уменьшается до того, которое может быть полностью реабсорбировано в канальцах. Поскольку механизм ацидификации в дистальных канальцах остается интактным, моча может подкисляться (рН менее 5,5). Поток через дистальные канальцы большого количества бикарбоната стимулирует реабсорбцию натрия в обмен на калий, что приводит к гипокалиемии. Уменьшение внеклеточного объема жидкости (в результате потери бикарбоната натрия) стимулирует реабсорбцию хлора (в результате чего возникает гиперхлоремия) и секрецию альдостерона (что приводит к увеличению потерь калия).
Проксимальный ПКА (таблица 15) может протекать как изолированно, не сочетаясь с другими заболеваниями, так и в сочетании с другими аномалиями функции проксимальных канальцев.
Изолированный проксимальный ПКА может быть транзиторным или персистирующим, спорадическим или наследственным (обычно аутосомно-доминантный тип). Проксимальный канальцевый ацидоз встречается и как составная часть генерализованного нарушения проксимального канальцевого транспорта (синдром Фанкони), характеризующийся глюкозурией, фосфатурией, аминоацидурией и проксимальным ПКА. Первичная форма синдрома Фанкони, также не связанная с другими формами заболевания, может наследоваться по аутосомно-доминантному и аутосомно-рецессивному типу. Вторичный синдром Фанкони может развиться при различных наследственных или приобретенных заболеваниях. Наследственные формы включают в себя перечисленные далее.
Цистинурия и цистиноз. Этот наследуемый по аутосомно-рецессивному типу дефект может определяться у детей первых 3 лет жизни (нефропатическая форма) и позднее (ювенильная форма). При нефропатической форме первоначальные клинические проявления заключаются в полиурии и полидипсии (нарушение концентрации), лихорадочном состоянии (дегидратация), задержке роста, рахите, у ребенка обычно белокурые волосы и светлая кожа (нарушение пигментации), он страдает фотофобией. Заболевание можно заподозрить при обнаружении кристаллов цистина в роговице при осмотре щелевой лампой, оно подтверждается по увеличению количества цистина в лейкоцитах. Накопление цистина в клетках почек приводит к прогрессирующему их повреждению с развитием терминальной почечной недостаточности к концу первого 10-летия жизни. Ювенильная форма заболевания проявляется позднее, она характеризуется теми же, но не столь тяжелыми проявлениями, хотя также может прогрессировать до развития почечной недостаточности.
Синдром Лоу. Этот синдром, сцепленный с Х-хромосомой, сопровождается отставанием умственного развития, гипотензией, катарактой, глаукомой и генерализованной дисфункцией проксимальных канальцев. Лежащий в основе метаболический дефект не установлен.
Галактоземия. Почечные проявления заболевания обусловлены накоплением галактозы в проксимальных канальцах.
Врожденная непереносимость фруктозы. Это аутосомно-рецессивное заболевание, сопровождающееся дефицитом фруктозо-1-фосфат-альдолазы, приводит к дисфункции проксимальных канальцев.
Тирозинемия. При наследственной форме тирозинемии часто развивается генерализованная дисфункция проксимальных канальцев.
Болезнь Вильсона. Клинические проявления этого аутосомно-рецессивого заболевания включают в себя проксимальную канальцевую дисфункцию.
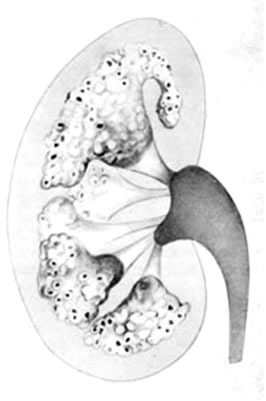
Медуллярная губчатая почка. Заболевание наследуется по аутосомно-доминантному типу, в то время как сходное с ним заболевание, ювенильный нефронофтиз, наследуется по аутосомно-рецессивному типу. Остается неясным, разные это заболевания или это - одно и то же заболевание с разными типами наследования. У детей чаще определяется рецессивный тип, у взрослых - доминантный тип. Важнейший патологоанатомический признак заключается в кистозе мозгового слоя. Поскольку кисты, как полагают, представляют собой расширение дистальных канальцев и собирательных трубок, часть из них может быть выявлены и в корковом слое. Прогрессирующее инстерстициальное воспаление и фиброз приводят к склерозу клубочков, атрофии коркового слоя и почечной недостаточности. Степень поражения пирамид может быть различной. Лишь в исключительных случаях заболевание ограничивается 1 - 2 пирамидами. Диаметр кист равняется 1 - 3 мм, редко достигает 6 мм. Кисты локализуются исключительно в зоне пирамид, не затрагивая кортикальный слой и бертиниевы столбы почки. Кисты иногда сообщаются с собирательными канальцами, а иногда с чашечкой. В полости кист часто обнаруживаются конкременты.
У некоторых детей клинически заболевание не проявляется до тех пор, пока не разовьется терминальная почечная недостаточность. У других развивается дисфункция канальцев, например полиурия и полидипсия (нарушение концентрационной способности почек), потеря натрия и проксимальный ПКА. Моча может быть в пределах нормы или в ней определяются минимальные изменения.
Диагноз ставится большей частью на основании экскреторной урографии, реже - ретроградной пиелографии. Экскреторная урография хорошо выявляет заполненные контрастным веществом гроздеподобные кисты в области сосочков и пирамид (рис. 25, 26). Иногда на обзорном снимке могут быть видны тени конкрементов с характерной их локализацией в дистальных отделах пирамид и неправильными контурами. Рентгенологическая картина годами может оставаться неизменной.
 |
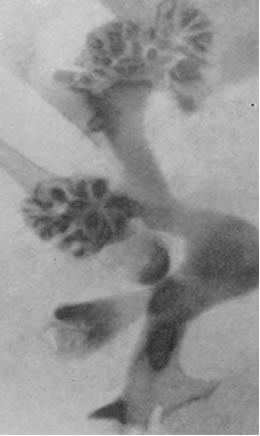 |
| Рис.25. Схема губчатой почки (Н.А. Лопаткин и соавт.). | Рис.26. Экскреторная урограмма больного с губчатой почкой и камнями в кистах (Н.А. Лопаткин и соавт.). |
Дифференциальный диагноз следует проводить со всеми заболеваниями, при которых наблюдаются поликистозные изменения в почечной паренхиме: поликистозными почками, мультилакулярной кистой, некротическим папиллитом, хроническим пиелонефритом и др.
Синдром Фанкони. К причинам приобретенного синдрома Фанкони относятся:
- воздействие канальцевых токсинов, таких как соли тяжелых металлов (свинец, ртуть, кадмий, уран), тетрациклин с истекшим сроком действия;
- состояния, сопровождающиеся протеинурией (миелома, нефротический синдром);
- интерстициальный нефрит.
Избыточная секреция паратгормона (первичный и вторичный гиперпаратиреоз, витамин D - дефицитный рахит) также может спровоцировать проксимальный ПКА (предположительно в результате ингибирования карбоангидразы).
98-103
| Назад | Вперед |